Pharmacophore Based Virtual Screening and Docking of Different Aryl Sulfonamide Derivatives of 5HT7R Antagonist
Article Sidebar
Main Article Content
Abstract
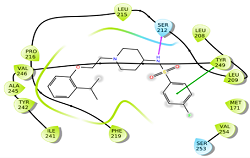
The selective blockade of 5HT7R (5-hydroxytryptamine 7 receptor) displays an antidepressant-like activity. It is a Gs-coupled receptor, which inactivates the adenyl cyclase enzyme or activates the potassium ion channel. Structural information of 5HT7 was obtained by homology modeling using MODELLER v.9.13. In the present study, pharmacophore-based virtual screening, molecular docking, and binding free energy calculations were performed on a series of antagonist aryl sulphonamide derivatives. A five-point pharmacophore hypothesis with two hydrogen bond acceptor (A), one hydrogen bond donor (D), one positive group (p), and one ring (R) was developed with acceptable R2 and Q2 values of 0.90 and 0.602, respectively. Eventually, common pharmacophore hypothesis-based screening was conducted against Asinex databases. Finally, binding free energy and dock score analysis was carried out for the top hits obtained from the docking process. All 14 hits from the database in this study had a satisfactory dock score and binding energy values within the best active compound range. H bond interaction with amino acid residues Ser212 and π-π stacking with Tyr249 were investigated for the best active molecule. Both are present in the top hits, including other interactions as well.
Downloads
Article Details

This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
Authors continue to retain the copyright to the article if the article is published in the Journal of Molecular Docking. They will also retain the publishing rights to the article without any restrictions.
Authors who publish with this journal agree to the following terms:
- Any article on the copyright is retained by the author(s).
- The author grants the journal, right of first publication with the work simultaneously licensed under a Creative Commons Attribution License that allows others to share work with an acknowledgment of the work authors and initial publications in this journal.
- Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of published articles of work (eg, post-institutional repository) or publish it in a book, with acknowledgment of its initial publication in this journal.
- Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their websites) prior to and during the submission process, as can lead to productive exchanges, as well as earlier and greater citation of published work.
- The article and any associated published material are distributed under the Creative Commons Attribution-ShareAlike 4.0 International License.




References
2. Ruat M, Traiffort E, Leurs R, Tardivel-Lacombe J, Diaz J, Arrang JM, Schwartz JC. Molecular cloning, characterization and localization of a high-affinity serotonin receptor (5-HT7) activating cAMP formation. Proc Natl Acad Sci U S A. 1993;90(18):8547-51. doi:10.1073/pnas.90.18.8547
3. Kvachnina E, Liu G, Dityatev A, Renner U, Dumuis A, Richter DW, et al. 5-HT7 receptor is coupled to Gα subunits of heterotrimeric G12-protein to regulate gene transcription and neuronal morphology. J Neurosci. 2005;25(34):7821-30. doi:10.1523/JNEUROSCI.1790-05.2005
4. Renner U, Zeug A, Woehler A, Niebert M, Dityatev A, Dityateva G, et al. Heterodimerization of serotonin receptors 5-HT1A and 5-HT7 differentially regulates receptor signalling and trafficking. J Cell Sci. 2012;125(10):2486-99. doi:10.1242/jcs.101337
5. Abbas AI, Hedlund PB, Huang XP, Tran TB, Meltzer HY, Roth BL. Amisulpride is a potent 5-HT 7 antagonist: relevance for antidepressant actions in vivo. Psychopharmacology. 2009;205(1):119-28. doi:10.1007/s00213-009-1521-8
6. Lajoie Y, Teasdale N, Bard C, Fleury M. Attentional demands for static and dynamic equilibrium. Exp Brain Res. 1993;97(1):139-44. doi:10.1186/s13065-018-0422-5
7. Pramanik S, Kutzner A, Heese K. 3D Structure, Dimerization Modeling, and Lead Discovery by Ligand‐protein Interaction Analysis of p60 Transcription Regulator Protein (p60TRP). Mol Inform. 2016;35(3‐4):99-108. doi:10.1186/s13065-018-0422-5
8. Pramanik S, Kutzner A, Heese K. Lead discovery and in silico 3D structure modeling of tumorigenic FAM72A (p17). Tumour Biol. 2015;36(1):239-49. doi:10.1186/s13065-018-0422-5
9. Zajdel P, Marciniec K, Maślankiewicz A, Satała G, Duszyńska B, Bojarski AJ, et al. Quinoline-and isoquinoline-sulfonamide derivatives of LCAP as potent CNS multi-receptor—5-HT1A/5-HT2A/5-HT7 and D2/D3/D4—agents: The synthesis and pharmacological evaluation. Bioorg Med Chem. 2012;20(4):1545-56. doi:10.1016/j.bmc.2011.12.039
10. Vrontaki E, Kolocouris A. Pharmacophore Generation and 3D-QSAR model development using PHASE. Methods Mol Biol. 2018;1824:387-401. doi:10.1007/978-1-4939-8630-9_23
11. Hughes JP, Rees S, Kalindjian SB, Philpott KL. Principles of early drug discovery. Br J Pharmacol. 2011;162(6):1239-49. doi:10.1111/j.1476-5381.2010.01127.x
12. Partyka A, Kurczab R, Canale V, Satała G, Marciniec K, Pasierb A, et al. The impact of the halogen bonding on D2 and 5-HT1A/5-HT7 receptor activity of azinesulfonamides of 4-[(2-ethyl) piperidinyl-1-yl] phenylpiperazines with antipsychotic and antidepressant properties. Bioorg Med Chem. 2017;25(14):3638-48. doi:10.1016/j.bmc.2017.04.046
13. Marciniec K, Kurczab R, Książek M, Bębenek E, Chrobak E, Satała G, et al. Structural determinants influencing halogen bonding: a case study on azinesulfonamide analogs of aripiprazole as 5-HT 1A, 5-HT 7, and D 2 receptor ligands. Chem Cent J. 2018;12(1):55. doi:10.1093/nar/gkm290
14. Kowalski P, Śliwa P, Satała G, Kurczab R, Bartos I, Zuchowicz K. The effect of carboxamide/sulfonamide replacement in arylpiperazinylalkyl derivatives on activity to serotonin and dopamine receptors. Arch Pharm. 2017;350(10):1700090. doi:10.1002/ardp.201700090
15. Canale V, Partyka A, Kurczab R, Krawczyk M, Kos T, Satała G, et al. Novel 5-HT7R antagonists, arylsulfonamide derivatives of (aryloxy) propyl piperidines: add-on effect to the antidepressant activity of SSRI and DRI, and pro-cognitive profile. Bioorg Med Chem. 2017;25(10):2789-99. doi:10.1016/j.bmc.2017.03.057
16. Li J, Abel R, Zhu K, Cao Y, Zhao S, Friesner RA. The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins. 2011;79(10):2794-812. doi:10.1002/prot.23106
17. Magrane M. UniProt Knowledgebase: a hub of integrated protein data. Database. 2011;2011:bar009. doi:10.1093/database/bar009
18. Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36(Suppl 2):W5-9. doi:10.1093/nar/gkn201
19. Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31(13):3497–500. doi:10.1093/nar/gkg500
20. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26(2):283–91. doi:10.1107/S0021889892009944
21. Eisenberg D, Lüthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997;277:396–404. doi:10.1016/s0076-6879(97)77022-8
22. Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–10. doi:10.1093/nar/gkm290
23. Kashif M, Hira SK, Upadhyaya A, Gupta U, Singh R, Paladhi A, et al. In silico studies and evaluation of antiparasitic role of a novel pyruvate phosphate dikinase inhibitor in Leishmania donovani infected macrophages. Int J Antimicrob Agents. 2019;53(4):508-14. doi:10.1016/j.ijantimicag.2018.12.011
24. Moussa N, Hassan A, Gharaghani S. Pharmacophore model, docking, QSAR, and molecular dynamics simulation studies of substituted cyclic imides and herbal medicines as COX-2 inhibitors. Heliyon. 2021;7(4):e06605. doi:10.1016/j.heliyon.2021.e06605
25. Peddi SR, Sivan SK, Manga V. Molecular dynamics and MM/GBSA-integrated protocol probing the correlation between biological activities and binding free energies of HIV-1 TAR RNA inhibitors. J Biomol Struct Dyn. 2018;36(2):486-503. doi:10.1080/07391102.2017.1281762
26. Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein− ligand complexes. J Med Chem. 2006;49(21):6177-96. doi:10.1021/jm051256o
27. Debnath T, Majumdar S, Kalle AM, Aparna V, Debnath S. Identification of potent histone deacetylase 8 inhibitors using pharmacophore-based virtual screening, three-dimensional quantitative structure–activity relationship, and docking study. Res Rep Med Chem. 2015;5:21-39. doi:10.2147/RRMC.S81388
28. Kaushik AC, Kumar S, Wei DQ, Sahi S. Structure Based Virtual Screening Studies to Identify Novel Potential Compounds for GPR142 and Their Relative Dynamic Analysis for Study of Type 2 Diabetes. Front Chem. 2018;6:23. doi:10.3389/fchem.2018.00023
29. Bhowmick S, Saha A, Osman SM, Alasmary FA, Almutairi TM, Islam MA. Structure-based identification of SARS-CoV-2 main protease inhibitors from anti-viral specific chemical libraries: an exhaustive computational screening approach. Mol Divers. 2021;25(3):1979-97. doi:10.1007/s11030-021-10214-6
30. Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10(5):449-61. doi:10.1517/17460441.2015.1032936
31. Rapp C, Kalyanaraman C, Schiffmiller A, Schoenbrun EL, Jacobson MP. A molecular mechanics approach to modeling protein-ligand interactions: relative binding affinities in congeneric series. J Chem Inf Model. 2011;51(9):2082-9. doi:10.1021/ci200033n
32. Mulakala C, Viswanadhan VN. Could MM-GBSA be accurate enough for calculation of absolute protein/ligand binding free energies? J Mol Graph Model. 2013;46:41-51. doi:10.1016/j.jmgm.2013.09.005
33. Kuchana V, Kashetti V, Peddi SK, Sivan S, Manga V. Integrated computational approach for in silico design of new purinyl pyridine derivatives as B-Raf kinase inhibitors. J Recept Signal Transduct Res. 2021:1-5. doi:10.1080/10799893.2021.1999472
34. Shehu Z, Uzairu A, Sagagi B. Quantitative structure activity relationship (QSAR) and molecular docking study of some pyrrolones antimalarial agents against plasmodium falciparum. J Turk Chem Soc Sect A Chem. 2018;5(2):569-84. doi:10.18596/jotcsa.346661
35. Le MT, Hoang VN, Nguyen DN, Bui THL, Phan TV, Huynh PNH, et al. Structure-Based Discovery of ABCG2 Inhibitors: A Homology Protein-Based Pharmacophore Modeling and Molecular Docking Approach. Molecules. 2021;26(11):3115. doi:10.3390/molecules26113115
36. Parker CG, Galmozzi A, Wang Y, Correia BE, Sasaki K, Joslyn CM, et al. Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell. 2017;168(3):527-41. doi:10.1016/j.cell.2016.12.029
37. Jorgensen WL, Duffy EM. Prediction of drug solubility from structure. Adv Drug Deliv Rev. 2002;54(3):355-66. doi:10.1016/s0169-409x(02)00008-x
38. Itteboina R, Ballu S, Sivan SK, Manga V. Molecular modeling-driven approach for identification of Janus kinase 1 inhibitors through 3D-QSAR, docking and molecular dynamics simulations. J Recept Signal Transduct Res. 2017;37(5):453-69. doi:10.1080/10799893.2017.1328442